摘要本文建立了不需要消化過程,以懸浮液進樣-石墨爐原子吸收法直接測定保健酒中鉛和錳的方法,研究 了加入穩定劑及基體改性劑對檢測結果的影響。結果表明,鉛、錳的相對標準偏差(RSD)分別為:Pb :2. 2%~ 2. 5% ,Mn :2.5%?2.9%。回收率分別為:Pb : 94%-105%; Mn :95%~106%。本法與國標法相比,具有操作 簡單、準確的優點。
隨著經濟的發展和生活水平的提高,人們對食品的食療功效越來越看重,保健酒因其保健功效為 廣大消費者青睞。然而在其生產過程中,由于加工 工藝及原料選取的不同,極易引起金屬元素超標,會對消費者的健康造成危害。目前國家標準中保健酒 鉛和錳的測定方法主要為原子吸收法。但該方 法不僅樣品前處理環節操作繁瑣,稍有不當還會造 成污染和被測元素損失。本文在參閱有關文獻研 究的基礎上,通過對丙三醇、三乙醇胺和草酸銨 3種穩定劑進行比較研究,建立了以丙三醇做為穩 定劑,加入合適的基體改進劑,對保健酒中的鉛和錳 采用直接進樣梯度升溫石墨爐原子吸收測定的方 法。與國標方法比較,本方法具有操作簡便、直 接、能有效避免污染和被測元素損失等優點,是 一種能夠用于保健酒中的鉛和錳進行快速準確分析 的方法。
I實驗部分
1.1材料與試劑
鉛、錳(以下簡稱:“兩元素”)標準儲備液 (lOOOμg/mL),三乙 醇胺(分析純)、丙三醇(分析純)、草酸銨(分析純)、 硝酸鎂(分析純), 硝酸(優級純)和二水合硝酸鈀(化學純)
1.2儀器與設備作者簡介:
原子吸收分光光度計,Pb原裝空心陰極 燈,Mn原裝空心陰極燈,Milli-QAlO超純水處理 系統(Millipore公司)0 1.3儀器工作條件 1.3.1 鉛 吸收線283. 3nm,工作電流8mA,狹縫寬度0. 7mn,測量類型吸收-背景,測量峰面積,進樣體積 20pL,基體改進劑IC^L,塞曼效應背景校正,測量 方式標準曲線法。 1.3.2 猛 吸收線279. 5nm,工作電流20mA,狹縫寬度 0. 2nm,測量類型吸收-背景,測量峰面積,進樣體積 20pL,基體改進劑IOmL,塞曼效應背景校正,測量 方式標準曲線法。 二元素石墨爐升溫程序見表I。
表I石蜃爐升溫程序
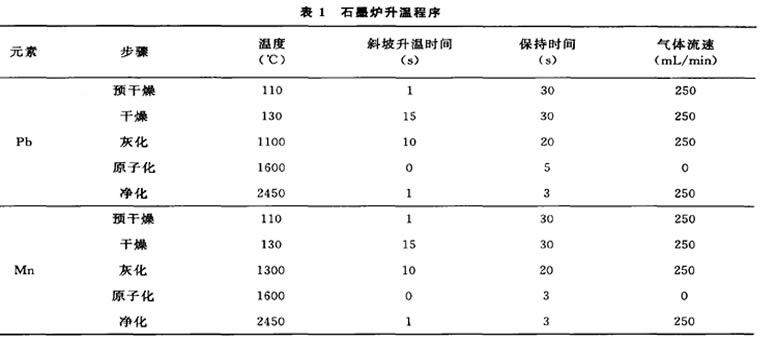
1.4試驗方法
基體改進劑的使用:在試樣測定的同時注人 1Oμl基體改進劑鈀-硝酸鎂溶液,以消除基體干 擾。繪制標準曲線與測定試劑空白時也應加人等量 的基體改進劑鈀-硝酸鎂溶液。
標準曲線制備:先將二元素標準儲備液分別準 確稀釋成為2OOpg/L的標準使用液,在測定時經由 自動進樣器再將上述標準使用液(200μg/L)分別自 動稀釋為以下濃度的標準系列:Pb: 0、25、50、75、 100μg/L;Mn:0、50、100、150、200μg/L。在檢測條 件下測定,自動繪制標準曲線。
直接進樣檢測:準確量取5. OmL混合均勻樣 品,加人5mL穩定劑,用超純水定容至25mL,混 勻。按檢測條件進行自動進樣測定。對于元素含量 超出標準工作曲線濃度范圍的樣品,可根據實際情 況定量稀釋后測定。
2結果分析 2.1懸浮液穩定劑的選擇
保健酒中本身允許含有少量的懸濁物質,如果 將樣品長時間靜置,可能會導致樣品不均勻,影響檢 測結果的穩定性。加人適量穩定劑,可以從很大程 度上減輕懸濁物質對于檢測結果的影響。一般常用 為穩定劑的有三乙醇胺[5]、丙三醇[4]、草酸銨[3]及乙 醇[6]等。本文選取了三乙醇胺、丙三醇和草酸銨分 別作為穩定劑進行實驗比較。表2比較了丙三醇
表2 3種穩定劑的結果比較 (n=3)
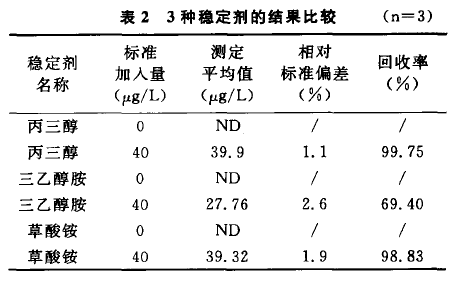
(4:1)),三乙醇胺(1:1))和草酸銨(0.007 mol/L)分別作為穩定劑時鉛的檢測結果。結果表 明添加丙三醇(4:1) (v/v)作為穩定劑效果*,回 收率可達到99. 75%,穩定性也(RSD% = I. I)。
2.2懸浮液穩定劑加入量的探討
準確量取5. OmL樣品6份,各加人I. 0、3. O、 5.0,6. 0,8. 0、10. OmL 丙三醇(4:l)(v/v),按樣品 進行測定。試驗表明,在樣品中加入大于5mL的丙 三醇(4:l)(v/v),就能實現很好的穩定(大于12小 時,已能滿足樣品測定所需的時間),且隨著丙三醇 (4:l)(v/v)的加入量增大,懸浮液穩定性也隨之增 加,但考慮到穩定劑的粘度過大,極易導致自動進樣 器進樣產生誤差,因而本文zui終將加入丙三醇(4:1) (v/v)的量確定為5mL。
2.3基體改性劑的選擇
Pb元素未加基體改進劑 Pb元素加入基體改進刑
基體改進劑能夠提高被測物的熱穩定性,使被 測元素能夠在較高的灰化溫度下不損失,從而達到 盡可能排除和減輕基體干擾的目的。本文分別試驗 比較了以下幾種基體改進劑:磷酸銨(O. 5g /L)、硝 酸鎂(O. 3g /L)、磷酸二氫銨(O. 5g /L)、磷酸二氫銨 (O. 5g /L)-硝酸鎂(O. 3g /L)混合溶液以及硝酸 (0.5g/L)_硝酸鎂(0.3g/L)混合溶液。試驗結果 表明,磷酸銨、硝酸鎂、磷酸二氫銨3種基體改進劑, 均可將灰化溫度提高至800'C左右,使用磷酸二氫 銨-硝酸鎂混合溶液可將灰化溫度提高至900X:左 右,而采用硝酸鈀-硝酸鎂混合溶液作為基體改進 劑,可將灰化溫度提高到1100'C以上。圖1、圖2分
400 600 800 1000 120() 1400 灰化溫度("C)
圖I鉛元素不同灰化溫度對加標回收率的影響
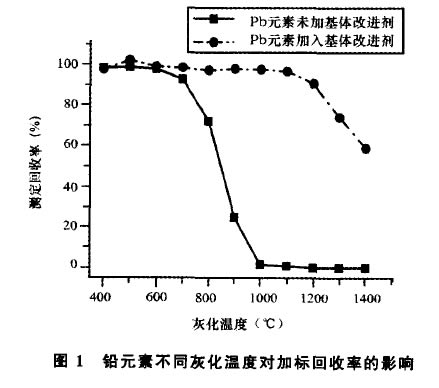
圖2錳元素不同灰化溫度對加標回收率的彩響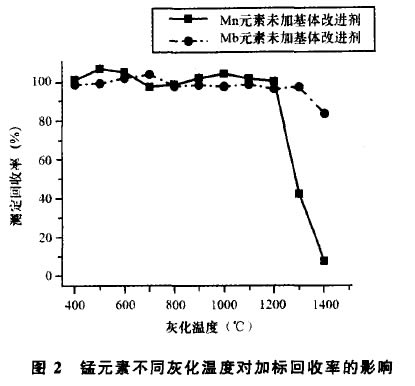
別為在不同灰化溫度下,添加與未添加硝酸鈀-硝 酸鎂混合溶液基體改進劑的二元素加標回收率曲線 圖。考慮到1ooo以上的灰化溫度能夠更為* 地消除保健酒中的復雜基體干擾,因此,本文zui終選 擇硝酸鈀(O. 5g /L)-硝酸鎂(0. 3g/L)混合溶液作 為基體改進劑。
2.4灰化溫度的選擇
灰化是石墨爐階梯升溫程序中的一個極其重要 的步驟,其目的是在被測元素不產生損失的前提下, zui大程度地消除基體組分對被測元素帶來的干擾。 由圖I可見,鉛元素在添加了硝酸鈀一硝酸鎂混合 溶液基體改進劑的情況下,在1100'C的灰化溫度仍 有較高的回收率,而looo'c以上,背景基體已基本 能夠揮發*,因此鉛的*灰化溫度定為 1100'c。錳元素的揮發溫度相比鉛元素則要高得 多,由圖2可見,在12001:的灰化溫度以下,添加與 未添加硝酸鈀-硝酸鎂混合溶液基體改進劑測得的 回收率差異并不是十分顯著。所以,為了能夠zui為 有效地發揮基體改進劑的效用,本文將錳的*灰 化溫度定為1300。
2.5準確度和精密度
按本方法將6種保健酒進行加標回收測定,二 元素的回收率分別為:Pb : 94%-105% ; Mn : 95%-106%。又對以上6種樣品的二元素分別作 6次平行測定,結果相對標準偏差(RSD)分別為: Pb:2.2%-2.5% ;Mn: 2.5%-2.9%。
2.6與國標方法的比較
分別應用本方法與國標方法中干灰化法[1’2]對 同一種保健酒樣品進行兩個梯度的加標回收,結果
見表3。二者進行t值檢驗,根據統計學處理表明, 前處理,具有操作簡便、直接、能有效避免污染 本方法與國標法相比無顯著性差異。且本方法無須 及被測元素損失等優點,有較好的借鑒意義。
表3兩種方法的鉛和錳測定結果比較 (n=6)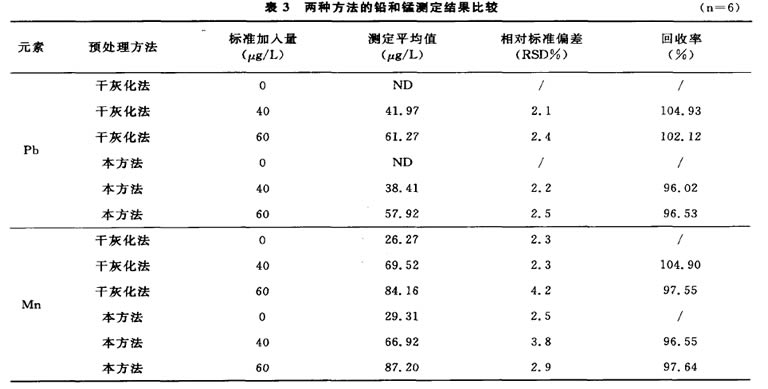
3結論
采用本方法測定保健酒中的鉛和錳,具有操作 簡便、直接等優點,適合針對批量樣品的準確測 定,具有很好的應用前景。
版權所有 © 2024 濟南精測電子科技有限公司 備案號:魯ICP備09052312號-5 技術支持:化工儀器網 管理登陸 GoogleSitemap
關鍵詞:氣相色譜儀、原子吸收分光光度計、液相色譜儀 、原子熒光



